
Requisitos de serialización para sector farmacéutico
En febrero de 2019 será de aplicación la serialización para el sector farmacéutico. A continuación, compartimos una guía sobre los requisitos requeridos y la forma de cumplir con ellos.
Los requisitos de serialización para el sector farmacéutico aseguran un proceso de verificación completo y efectivo y tienen el objetivo de eliminar la posibilidad de que ingresen medicamentos falsificados al mercado.
Para ello, a partir de la Directiva 2011/62/UE que modifica la Directiva 2001/83/CE sobre medicamentos de uso humano, los fabricantes de medicamentos deben incorporar en los envases de los fármacos características que garanticen la seguridad del producto y eviten la falsificación.
Las disposiciones detalladas relativas a los dispositivos de seguridad a incluir en los envases de los medicamentos de uso humano se recogen en el Reglamento Delegado (UE) 2016/161 de la Comisión.
La gran novedad acerca de este proceso es la implementación de un Registro de Información Digitalizado que obliga al uso de un Código Único de Identificación impreso en el embalaje de cada medicamento que además deberá contar con un tamper-evident -evidencia de no manipulación- que garanticen la autenticación y verificación de extremo a extremo, de todos los medicamentos.
Dada la importancia de esta regulación (las empresas que no den cumplimiento a los requisitos de serialización para el sector farmacéutico no podrán comercializar medicamentos en la Unión Europea) y que será de aplicación próximamente, el 9 de febrero de 2019, a continuación, desgranamos cuáles son los requisitos de serialización para el sector farmacéutico y qué se exige en cada fase del proceso de producción, distribución y comercialización.
Requisitos de serialización para sector farmacéutico
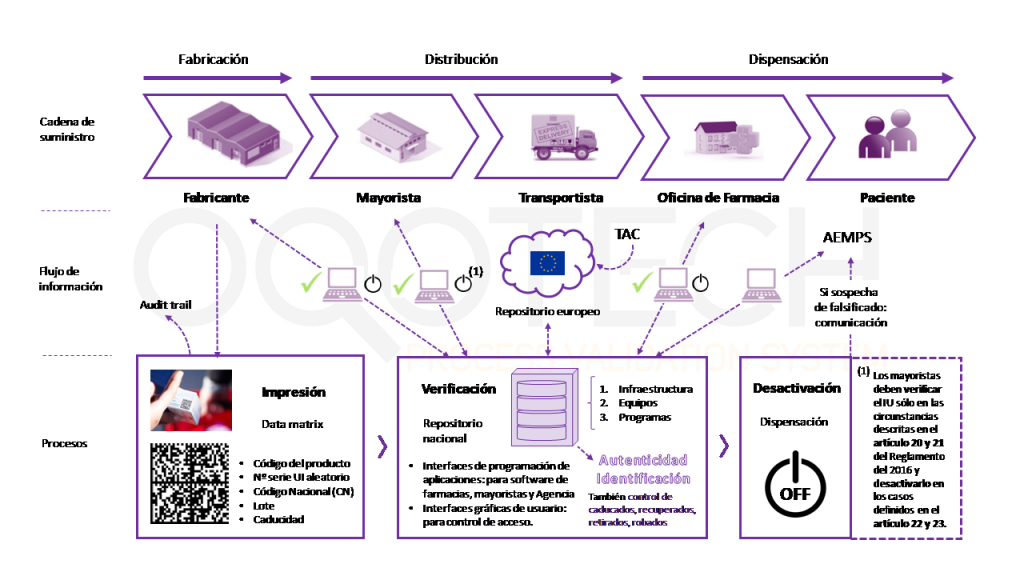
Los requisitos de serialización para el sector farmacéutico tienen aplicación a lo largo de toda la cadena de valor: producción (Fabricante, Titular de la autorización), distribución (Mayoristas) y dispensación (Personas autorizadas o facultadas para dispensar medicamentos).
A grandes rasgos, el proceso, como vemos en la imagen de debajo es el siguiente:
- Los fabricantes, los mayoristas y las personas autorizadas o facultadas para dispensar medicamentos verificarán la autenticidad del identificador único comparándolo con los identificadores únicos almacenados en el sistema de repositorios, así como la integridad del dispositivo contra las manipulaciones.
- Los fabricantes son responsables de generar e incluir en cada medicamento el IU (identificador único) así como un cierre “antiviolable”, y conservar la trazabilidad del proceso (audit trail).
- El titular de la autorización de comercialización o la persona responsable de la comercialización es responsable de enviar la información correspondiente al organismo nacional de verificación (SEVeM) para que forme parte del repositorio nacional que a su vez conectará con el repositorio europeo. Y, por tanto, de garantizar la introducción de la información en el sistema de repositorios.
- Los mayoristas deben verificar el IU (en las circunstancias descritas en los artículos 20 y 21 del Reglamento Delegado (UE) 2016/161) y desactivarlo en los casos definidos en los artículos 22 y 23.
Identificador único – Especificaciones técnicas
El laboratorio tiene la obligación de estampar en los envases de todos los medicamentos un identificador único de acuerdo con las siguientes especificaciones:
a) consistirá en una secuencia de caracteres alfanuméricos o sólo numéricos única para cada envase.
b) constará de:
- El código del producto debe identificar el nombre del producto, su denominación común, la dosis, la forma farmacéutica, el tipo de envase y el tamaño.
- El identificador único también incorporará una secuencia alfanumérica o solo numérica, que conste de no más de 20 caracteres – generados por algoritmos aleatorio determinista o no determinista – utilizando una estructura y sintaxis normalizadas de los datos.
- Si el medicamento tiene como destino un estado miembro de la Unión, que solicite un número de identificación nacional (CN), este debe estar estampado en el envase.
- Número de lote.
- Fecha de caducidad.
- La secuencia de caracteres numéricos o alfanuméricos utilizados para construir el identificador único serán exclusivos para el envase utilizado, hasta un año después de la fecha de caducidad del medicamento, o hasta cinco años después de que el medicamento haya sido puesto a la venta o distribuido (si este plazo fuera mayor).
- La estructura y calidad de impresión del código de barras bidimensional que contenga el identificador único deben permitir su lectura en alta velocidad y minimizar errores de lectura.
- Los elementos de datos del identificador único deben estar impresos en formato legible por las personas, para que pueda verificarse la autenticidad del identificador único y desactivarse este en caso de que el código de barras bidimensional sea ilegible.
Incorporar el identificador único al envase
Para efectos del cumplimiento de los requisitos de serialización para el sector farmacéutico, el fabricante codifica el identificador único, utilizando un código de barras bidimensional.
Este código de barras debe ser una matriz de datos que puede ser leída con instrumentos de lectura óptica, que permiten detección de errores, y corrección de los mismos. Para ello, el código de barras es conveniente que se ajuste a la norma ISO/IEC 16022:2006.
En cuanto a la calidad de impresión, el fabricante debe garantizar que sea suficiente para garantizar la lectura hasta al menos un año después de la fecha de caducidad del medicamento, o hasta cinco años después de que el éste haya sido puesto a la venta o distribución (si este plazo fuera mayor). Para el cumplimiento de este apartado del reglamento nos puede ser de utilidad la norma ISO/IEC 15415:2011, siendo la calidad de impresión como mínimo de 1,5 (nivel mínimo ISO).
Sistema de repositorios
Los repositorios pueden ser nacionales (de un estado miembro) o supranacionales (de varios estados miembro).
Cada estado miembro dispondrá del servicio de un repositorio nacional o supranacional. Dicho repositorio estará físicamente en el territorio de la Unión y podrá interoperar con los demás repositorios del sistema.
El sistema de repositorios deberá tener la infraestructura, equipos y programas informáticos necesarios para poder realizar las funciones de carga, almacenamiento y modificación de información, así como la identificación de dispositivos de seguridad, verificación de la autenticidad del IU y desactivación.
En cuanto a las especificaciones técnicas para llevar a cabo la verificación, el sistema de repositorios nacional debe contener:
- Plataforma central de información y datos, estando los repositorios conectados a dicha plataforma.
- Interfaces de programación de aplicaciones: para software de farmacias, mayoristas y Agencia nacional.
- Interfaces gráficas de usuario: para control de acceso.
Desactivación de medicamentos
De acuerdo al Reglamento Delegado (UE) 2016/161, el principio a seguir es que la desactivación del IU en el sistema de repositorios se ha de realizar cuando se dispensa el medicamento, es decir, al final de la cadena de suministro. Sin embargo, también es necesario garantizar la desactivación en cualquier otro punto de la cadena de suministro para aquellos envases que no puedan llegar a dispensarse (por ejemplo: si se distribuyen fuera de la UE, muestras solicitadas por las autoridades competentes, etc. ).
De acuerdo con lo anterior, no podrán ser distribuidos, comercializados o dispensados, medicamentos cuyo identificador único haya sido desactivado. Por supuesto, existen algunas excepciones y las particularidades sobre la verificación del identificador único se pueden consultar en el artículo 12 del Reglamento Delegado (UE) 2016/161.
Si en cualquier etapa de la cadena de suministro, el fabricante, el mayorista o la farmacia presumen que un medicamento ha sido manipulado o es falso, no lo pondrá a la venta, e informará de inmediato a la autoridad competente (AEMPS).
Desde Oqotech contamos con más de 10 años de experiencia informatizando procesos y validando sistemas informatizados en el sector farmacéutico y podemos ayudarte con todo lo necesario para dar conformidad a los requisitos de serialización. Contacta con uno de nuestros consultores expertos.







